CryoNet.Refine: A One-step Diffusion Model for Rapid Refinement of Structural Models with Cryo-EM Density Map Restraints
{kind=link}
Abstract
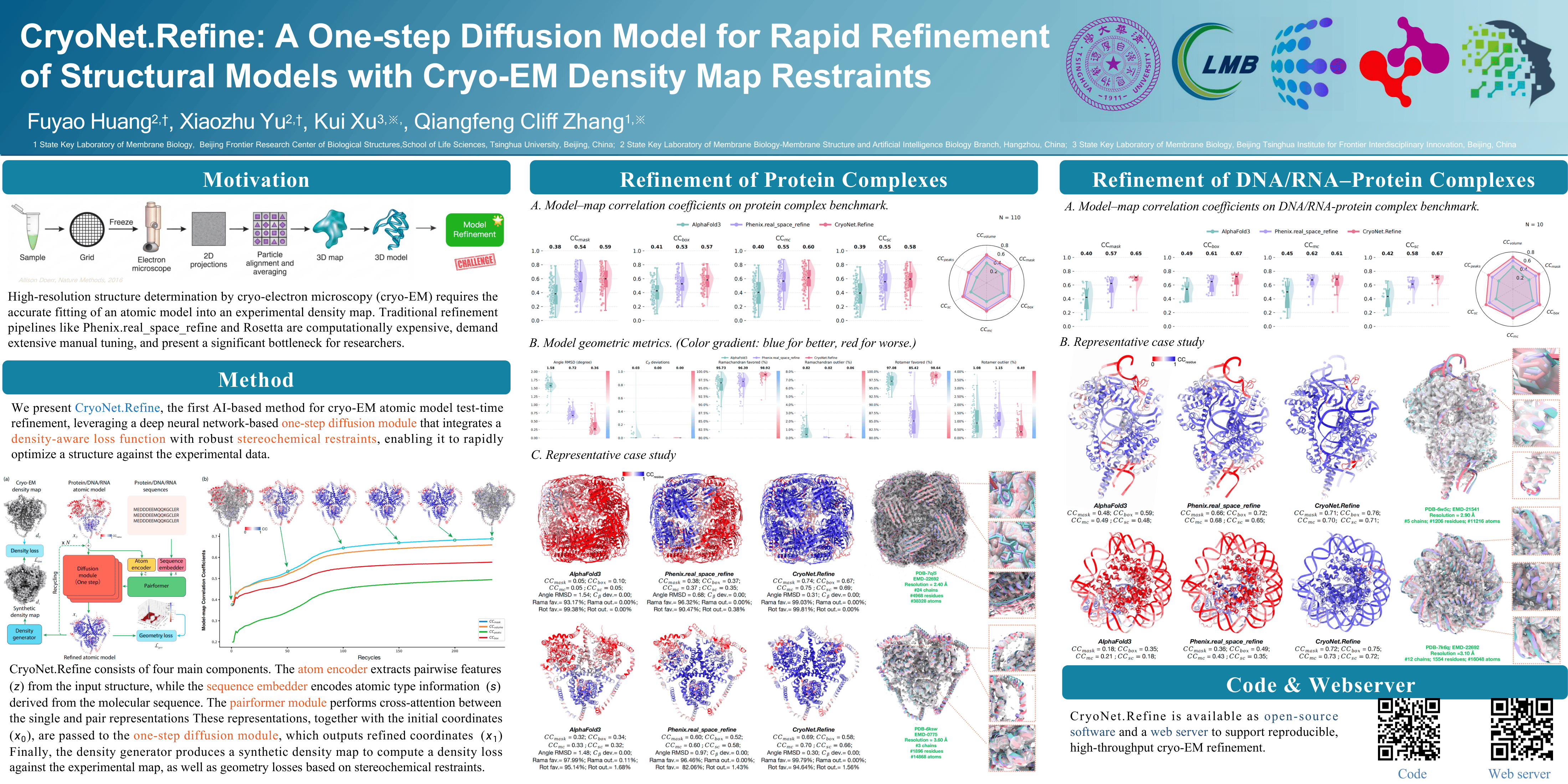
High-resolution structure determination by cryo-electron microscopy (cryo-EM) requires the accurate fitting of an atomic model into an experimental density map. Traditional refinement pipelines like Phenix.realspacerefine and Rosetta are computationally expensive, demand extensive manual tuning, and present a significant bottleneck for researchers. We present CryoNet.Refine, an end-to-end, deep learning framework that automates and accelerates molecular structure refinement. Our approach utilizes a one-step diffusion model that integrates a density-aware loss function with robust stereochemical restraints, enabling it to rapidly optimize a structure against the experimental data. CryoNet.Refine stands as a unified and versatile solution capable of refining not only protein complexes but also nucleic acids (DNA/RNA) and their assemblies. In benchmarks against Phenix.realspacerefine, CryoNet.Refine consistently yields substantial improvements in both model–map correlation and overall model geometric quality. By offering a scalable, automated, and powerful alternative, CryoNet.Refine is poised to become an essential tool for next-generation cryo-EM structure refinement. Web server: https://cryonet.ai/refine; Source code: https://github.com/kuixu/cryonet.refine.