Latent Conservative Objective Models for Offline Data-Driven Crystal Structure Prediction
{kind=link}
Abstract
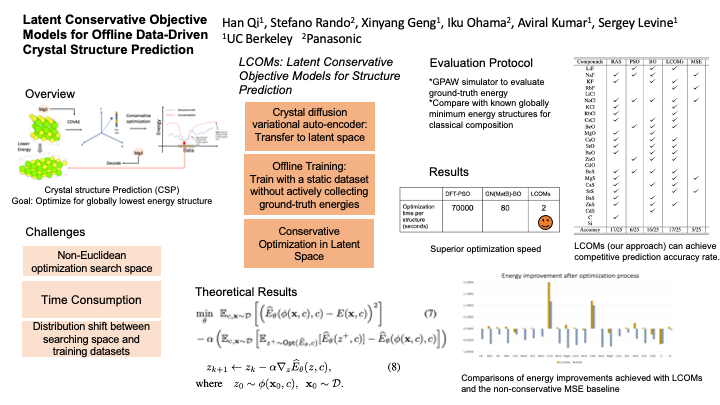
In computational chemistry, crystal structure prediction (CSP) is an optimization problem that involves discovering new crystal structures. This problem is challenging for machine learning (ML) methods: it requires discovering globally optimal designs that attain the smallest energy in complex non-Euclidean manifolds. One approach to tackle this problem involves building simulators based on density functional theory (DFT), but these simulators are painfully slow. More recent approaches are exploring the alternate paradigm of relying on learned graph neural networks (GNNs) surrogate models as a proxy for simulation. We propose a method that leverages GNNs to reduce the complexity of the problem. Concretely, we reduce the non-Euclidean optimization search space to a standard vector one with Graph Variational Autoencoders (GVAEs), and we combine that with techniques from offline model-based optimization. This prevents the optimization procedure from producing unstable structures that erroneously appear to have low energies under the learned model. We show that this procedure outperforms current alternatives, both in terms of success rate of structure prediction, and computational cost. In addition, it provides a generic recipe to apply offline optimization techniques for optimizing in non-Euclidean spaces.