CrysGNN : Distilling pre-trained knowledge to enhance property prediction for crystalline materials.
{kind=link}
Abstract
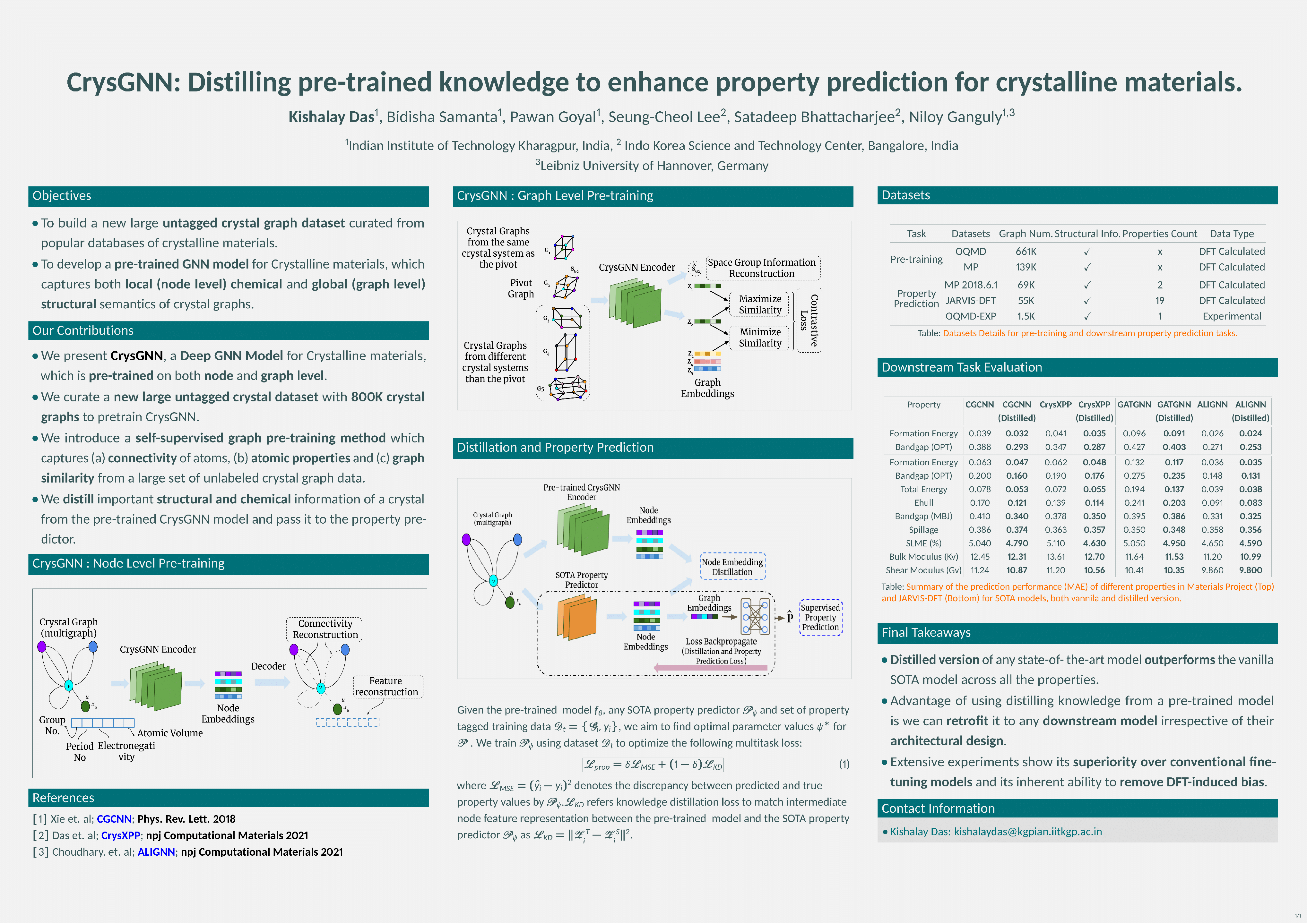
In recent years, graph neural network (GNN) based approaches have emerged as apowerful technique to encode complex topological structure of crystal materials inan enriched representation space. These models are often supervised in nature andusing the property-specific training data, learn relationship between crystal structureand different properties like formation energy, bandgap, bulk modulus, etc. Mostof these methods require a huge amount of property-tagged data to train the systemwhich may not be available for different properties. However, there is an availabilityof a huge amount of crystal data with its chemical composition and structural bonds.To leverage these untapped data, this paper presents CrysGNN, a new pre-trainedGNN framework for crystalline materials, which captures both node and graphlevel structural information of crystal graphs using a huge amount of unlabelledmaterial data. Further, we extract distilled knowledge from CrysGNN and injectinto different state of the art property predictors to enhance their property predictionaccuracy. We conduct extensive experiments to show that with distilled knowledgefrom the pre-trained model, all the SOTA algorithms are able to outperform theirown vanilla version with good margins. We also observe that the distillation processprovides a significant improvement over the conventional approach of finetuningthe pre-trained model.