Boltzmann-Aligned Inverse Folding Model as a Predictor of Mutational Effects on Protein-Protein Interactions
Xiaoran Jiao ⋅ Weian Mao ⋅ Wengong Jin ⋅ Peiyuan Yang ⋅ Hao Chen ⋅ Chunhua Shen
2025 Poster
{kind=link}
Abstract
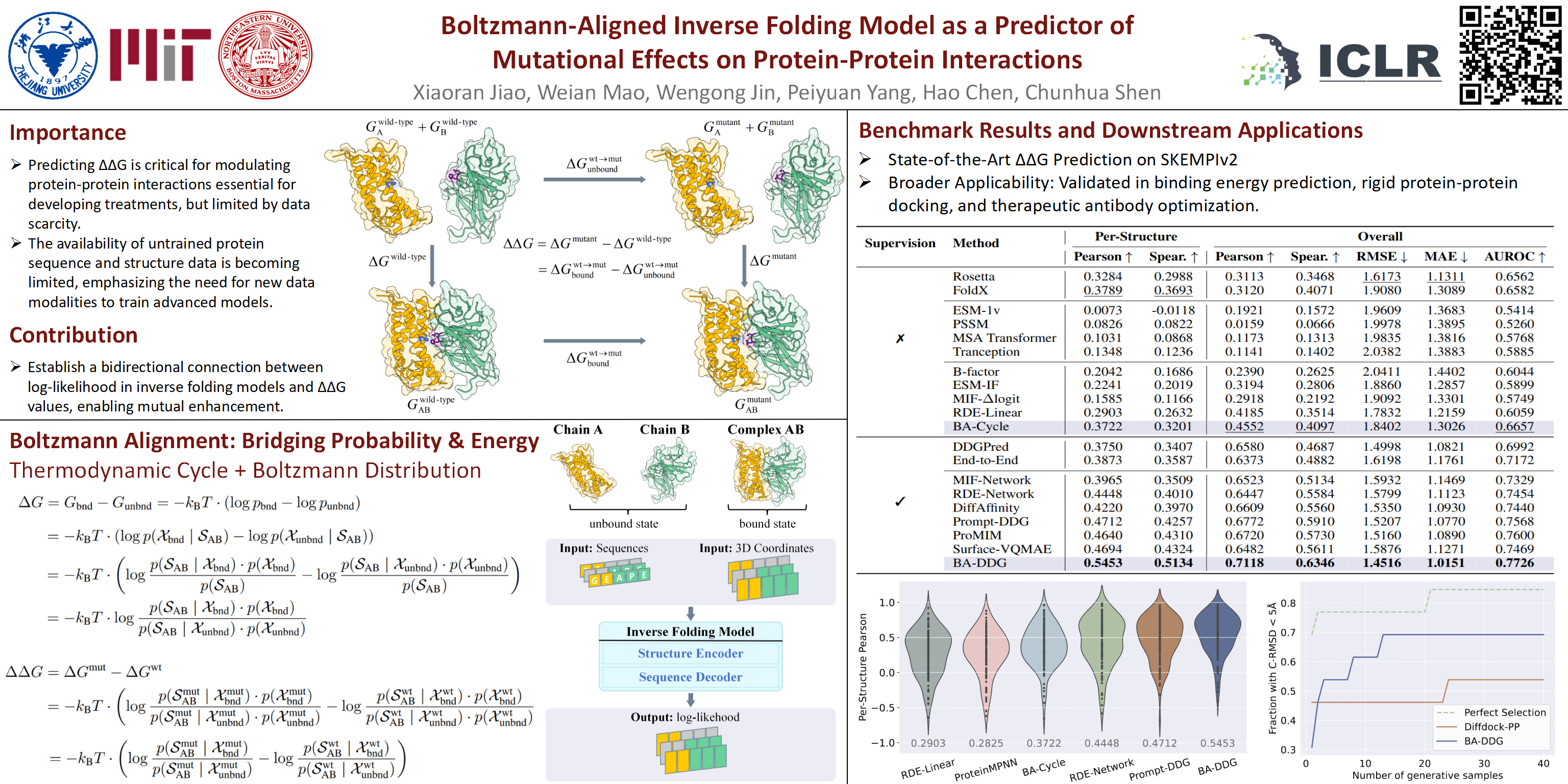
Predicting the change in binding free energy ($\Delta \Delta G$) is crucial for understanding and modulating protein-protein interactions, which are critical in drug design.Due to the scarcity of experimental $\Delta\Delta G$ data, existing methods focus on pre-training, while neglecting the importance of alignment.In this work, we propose Boltzmann Alignment technique to transfer knowledge from pre-trained inverse folding models to prediction of $\Delta\Delta G$.We begin by analyzing the thermodynamic definition of $\Delta\Delta G$ and introducing the Boltzmann distribution to connect energy to the protein conformational distribution. However, the protein conformational distribution is intractable. Therefore, we employ Bayes’ theorem to circumvent direct estimation and instead utilize the log-likelihood provided by protein inverse folding models for the estimation of $\Delta\Delta G$. Compared to previous methods based on inverse folding, our method explicitly accounts for the unbound state of the protein complex in the $\Delta \Delta G$ thermodynamic cycle, introducing a physical inductive bias and achieving supervised and unsupervised state-of-the-art (SoTA) performance.Experimental results on SKEMPI v2 indicate that our method achieves Spearman coefficients of 0.3201 (unsupervised) and 0.5134 (supervised) on SKEMPI v2, significantly surpassing the previously reported %SoTA valuesSoTA results of 0.2632 and 0.4324, respectively.Furthermore, we demonstrate the capability of our method in bindingenergy prediction, protein-protein docking, and antibody optimization tasks.Code is available at [https://github.com/aim-uofa/BA-DDG](https://github.com/aim-uofa/BA-DDG)
Video
Chat is not available.
Successful Page Load